To Extract the Values from an Easily Interpreted (First-Order) 1-H Spectrum:

Process your spectrum and Reference the frequency scale.
If you change the scale reference after extracting the chemical shifts, they will remain non corrected!
Define the integral regions
and normalize the integrals: the area values should be equal to the number
of protons that each signal arises from.
Perform the Peak-Picking.
It's important to select the correct number of peaks.
For example, a doublet of triplets consists of 6 peaks. If you have picked any extra-peak,
for example an impurity, delete that label.
When a coupling constant is small, it is necessary to resort to a resolution enhancement,
otherwise the peak-picking cannot find all the lines. After you have extracted the couplings,
you can remove peak-picking and/or the resolution enhancement without affecting the extracted values.
If the peaks are resolved, but the multiplets are not separated,
you should perform a selective peak-picking for each multiplet before point 4 below and
delete it before before going to point 5.
Choose View > J Couplings. This command opens the J Manager for the current document.
Select a multiplet. Press the key E (lowercase) or click the extract icon. A new entry appears into the J Manager. Change the name (first column) if you like. The same name appears on the plot, above the same multiplet.
Repeat for all the multiplets and fill the table. If a multiplet can't be resolved, only the chemical shift range will be reported.
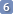
Check for consistency. Two coupling partners have, in theory, an identical constant. You can keep any experimental discrepancy, or overwrite the value that you think is less reliable with the constant measured from the partner. The table is editable.
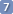
To substitute two inconsistent couplings with their average value, select the corresponding rows, then click on Match. To undo press TWICE the icon undo.
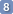
When you click Report, a preview of the list appears at the bottom of the J Manager. You can select a different reporting style with the icon Style. If you don't know which one to choose, you can try them all. When you have set a style for proton spectra and another style for heteronuclear spectra, they will remain in memory forever.
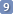
What you see is already on the clipboard. You can paste it into a Word document, for example.

To reproduce the same information, graphically, on the plot, click graph.