
step by step instructions to
Simulate Chemical Exchange with iNMR
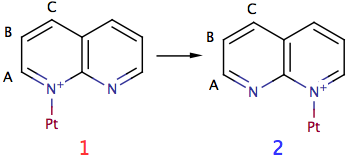
The picture shows a portion of a metallic complex. The ligand is bi-dentate and symmetric. The reaction consists into the breaking of a N-Pt bond and the creation of another one. The starting and final structures are identical. The hydrogen marked as A, however, moves away from platinum and as a consequence its chemical shift changes (and the Pt-H coupling disappears). A similar thing happens to hydrogens B and C. We don't need to worry about nuclei D, E and F in formula (1) because they are just the same as A, B and C of (2). For the same reason, we'll ignore D, E and F in formula (2) (they are the same as A, B and C of (1)).

Platinum is a mixture of 5 isotopes. Only one is visible in NMR: 195Pt, with an abundance of 33.8% and spin = 1/2. iNMR, when simulates chemical exchange, can handle one kind of nuclei only. To overcome the limitation, we'll declare the platinum as a fourth hydrogen (D), with a far away shift. We should declare, along this line, the two states in the figure, with 3 hydrogens each and abundance 66.2%, and another couple of states, with abundance 33.8% and 4 nuclei. There's another limitation of the program, however: all states must have the same number of nuclei. We'll declare, therefore, 4 nuclei for all states. Where the platinum isotope is non active, we'll set its coupling constants to zero. In our complex, only hydrogen A is coupled with Pt.
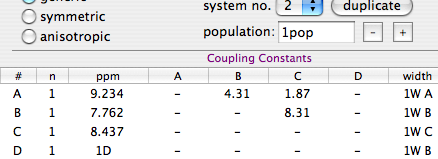
First we declare the four states to the program.
Create a new document with Cmd-N.
Uncheck the option “same width for all lines”.
Fill the table with the values shown here above. They are taken from a spectrum at very low temperature,
in the absence of exchange.
Press the button “duplicate” to create system no. 2.
Change the values as shown by the snapshot at the right.
Note that we only declare 3 independent line-width values (the numerical values).
Literal values like
“W B”, “1D”, “1pop”, etc... are references to already declared parameters.
From the menu, select system no. 1 and duplicate it again (no. 3 is created).
Duplicate system no. 2 too (no. 4 is created). Enter the values here below:

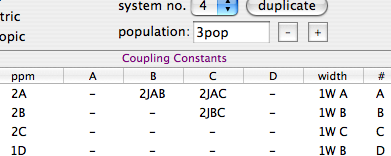
The chemical shifts and the Js of the new systems, characterized by the presence of 195Pt, are linked to those already defined. The population is the half, according to the isotopic composition. The signal of A in the third system is quite broad and coupled to Pt (J = 30 Hz).
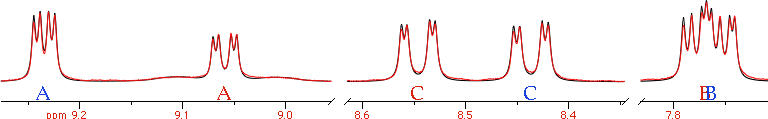
You know the parameters because they are already reported here. Normally you enter approximate values, then you refine them by direct comparison with the spectrum. The supplemental material to this article includes both the experimental spectra and their theoretic counterpart. Open the file sim184.spins, which already contains all the parameters and the corresponding experimental spectrum (red overlay). When you overlay the experimental spectrum, remember to click the big square button called “fix”. In this way the spectrometer frequency value is imported into the simulation. You can change a parameter by editing the value, or clicking the small arrows or, in the case of chemical shifts, by dragging the red and blue labels.
You'll simulate the other spectra in order of increasing temperature. Instead of defining the systems each time, you duplicate the previous simulation and correct the parameters as needed. To introduce the exchange, issue the commannd “Simulate/Dynamic”. iNMR creates 6 adjustable rate constants. Most of them will remain unused. For example, the system 1 cannot be directly transformed into 3 and, in any case, the transformation is so slow (compared to the intra-molecular rearrangement) that the corresponding constant can remain set to zero. It's important to set k34 = k12. At this point, we have informed the program that the process is governed by a single rate constant.
When you manually fit a spectrum, first adjust the chemical shifts. Before coalescence you can easily drag the individual red and blue letters. Near and after coalescence, this becomes unsafe. Turn on the option “Lock Equal Letters”. It locks the difference between two exchanging shifts. You can also adjust the Js at low temperatures, although they change little upon heating or cooling.
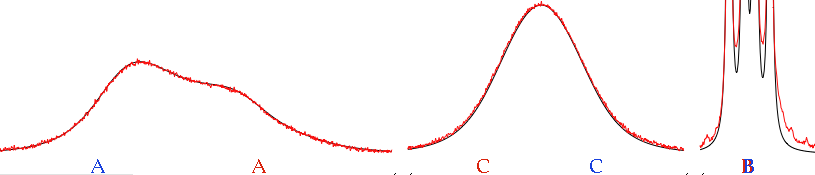
The shape of peaks near coalescence is very sensitive to the rate constant. When changing it, aim to fit the peaks that coalesce at that temperature. Only after you have fixed the rate constant, you can change the transversal relaxation times (line-widths). Not before, because relaxation and exchange have similar effects on the spectrum. If you don't know the rate of exchange, you risk to simulate through relaxation the broadening caused by exchange. The problem doesn't exist near coalescence, because the apparent width, in this case, comes almost completely from exchange.
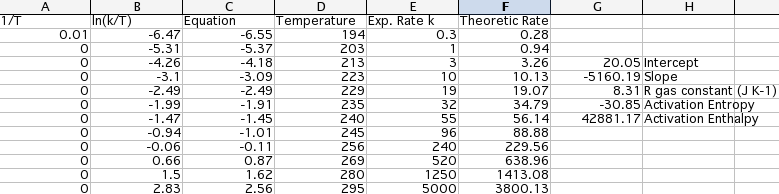

Each simulated spectrum furnishes a couple of values (temperature and rate of exchange). The supplemental material contains a spreadsheet in Open Office format (Eyring.ods) and its translation for Excel. The temperatures have been inserted into column D, the rates into column E. The Eyring equation gives the enthalpy and entropy of activation. The corresponding plot gives a visual indication of the correctness of the study. It is limited, however, to a single parameter (the most important one, but not the only one). You must verify with other means that all the parameters change in a regular way with temperature.
iNMR has tightly integrated established theory with a user-friendly interface. If you want to use iNMR for your own simulations, remember to read the manual. The contents of this article were also the subject of a seminar.
Disclaimer
There is no warranty. No one has reviewed the supplemental material.
No one has verified the formulas in the spreadsheets.
No one has certified the conformity of iNMR to any particular activity.
In other words: There is no warranty.
You are free to perform all those validations by yourself.